Showing posts with label biosynthesis. Show all posts
Showing posts with label biosynthesis. Show all posts
Sunday, 31 December 2023
A possible BioB bipass route
Nearly a decade ago there was something bugging me and I believe I have figured it out —although it's pointless now. Namely, is another way of making biotin possible without using BioB, biotin synthase, an incredibly slow multistep radical SAM enzyme.
Thursday, 16 May 2019
The secondary metabolism of pineberry strawberries

Saturday, 12 September 2015
Unnatural amino acid biosynthesis
In the synthetic biology experiments with an expanded genetic code the biosynthesis of unnatural amino acid is not taken into consideration as the system is rather rickety and the amino acids unusual.
Similar amino acids
A lot of introduced amino acids are dramatically different from the standard set. However, there are several amino acids that never made it to the final version of the genetic code as they are subtly different from the canonical amino acids and must have been too hard for the high promiscuous primordial systems to differentiate. However, subtle differences would be useful for finetuning. Examples include aminobutyric acid (homoalanine), norvaline and norleucine, allo-threonine ("allonine"), ornithine or aminoadipate. If there was a way to introduce them and increase the fidelity it would be hugely beneficial. It would probably result in enzymes with higher fidelity and catalytic efficiency.
That Nature itself failed back then does not necessary mean that scientists would fail with modern metabolism. The main drawback is a selection system. Current approached to recoding rely on the new amino acid as fill in as opposed to something that makes the E. coli addicted to it. The latter would mean that the system could evolve to better handle the new amino acids. Phage with an unnatural amino acid have higher fitness (Hammerling et al., 2014). Unnatural RNA display (Josephson et al., 2005) could be used to generate a protein that is evolved to require the non-canonical amino acid as nearby residues are evolved to best suit that enzyme, if one really wanted to all that trouble. Alternatively and less reliably, GFP with different residues does behave differently and position 65 could handle stuff like homoalanine, but the properties between S65A or S65V are not too different. So if a good and simple selection method were present it could be doable.
Most of the amino acids that nature can make would just make structural variants. Furthermore, mechanistic diversity mostly comes from cofactors (metals, PLP, biotin, thiamine, MoCo, FeS clusters etc.), which is a more sensible solution given that there only one per certain type of enzyme. Some amino acids that are supplemented Nature cannot make with ease, in particular chlorination and fluorination reactions in Nature can be counted with one hand.

The simplest novel amino acid is homoalanine (aminobutyrate). 2-ketobutyrate is produced during isoleucine biosynthesis, which if it were transaminated it would produce homoalanine. Therefore it is likely that the branched chain transaminase probably must go to some effort to not produce homoalanine (forbidden reaction). This amino acid is found in meteorites and is really simple, but its similarity to alanine, hence why it must have lost out.
 While on the topic, the whole route for phenylalanine biosynthesis is odd. It feels like an evolutionary remnant. If one were to draw up phenylalanine biosynthesis without knowing about the shikimate/chorismate pathway the solution would be different. If I were to design the phenylalanine pathway I would start with a tetraketide (terminal acetyl-CoA derived; 2,4,6,8-tetraoxononanoyl-CoA), cyclise (Aldol addition of C9 in enol form to C4 ketone), two rounds of reduction (6-oxo and 8-oxo) and three dehydrations (4,6,8-hydroxyl), followed by a transamination (2-oxo): phenylalanine by polyketide synthesis!
While on the topic, the whole route for phenylalanine biosynthesis is odd. It feels like an evolutionary remnant. If one were to draw up phenylalanine biosynthesis without knowing about the shikimate/chorismate pathway the solution would be different. If I were to design the phenylalanine pathway I would start with a tetraketide (terminal acetyl-CoA derived; 2,4,6,8-tetraoxononanoyl-CoA), cyclise (Aldol addition of C9 in enol form to C4 ketone), two rounds of reduction (6-oxo and 8-oxo) and three dehydrations (4,6,8-hydroxyl), followed by a transamination (2-oxo): phenylalanine by polyketide synthesis!

Phenylalanine has a single aromatic ring, naphthylalanine has two. Using the logic for the rethought phenylalanine synthesis we get a synthesis by hexaketide (2,4,6,8,10,12-hexaoxotridecanoyl-CoA). Namely cyclise (Aldol addition of C11 in enol form to C6 ketone and C13 to C4), two rounds of reduction (8-oxo and 10 or 12-oxo) and five dehydrations (4,6,8,10,12-hydroxyl), followed by a transamination (2-oxo). Naphthylalanine has been added to the genetic code (Wang et al., 2002), but was added as a supplement. GFP doesn’t work with either form of naphthylalanine (Kajihara et al., 2005), therefore there isn’t a good selection marker where the amino acid itself is beneficial. So probably the worst sketched pathway to possibly make.
That Nature itself failed back then does not necessary mean that scientists would fail with modern metabolism. The main drawback is a selection system. Current approached to recoding rely on the new amino acid as fill in as opposed to something that makes the E. coli addicted to it. The latter would mean that the system could evolve to better handle the new amino acids. Phage with an unnatural amino acid have higher fitness (Hammerling et al., 2014). Unnatural RNA display (Josephson et al., 2005) could be used to generate a protein that is evolved to require the non-canonical amino acid as nearby residues are evolved to best suit that enzyme, if one really wanted to all that trouble. Alternatively and less reliably, GFP with different residues does behave differently and position 65 could handle stuff like homoalanine, but the properties between S65A or S65V are not too different. So if a good and simple selection method were present it could be doable.
Novel amino acid biosynthesis
This leaves with the biosynthesis of the novel amino acids, which is the main focus here. There are many possible amino acids to choose from, and a good source of information for that is the wikipedia article non-proteinogenenic amino acids — I (reticently) wrote many years ago to sort out the mess that there was, but I subsequently left to the elements and it has become a bit cluttered like an unselected psuedogene. Why certain amino acids made it while other did not is discussed in a great paper by Weber and Miller in 1981.Most of the amino acids that nature can make would just make structural variants. Furthermore, mechanistic diversity mostly comes from cofactors (metals, PLP, biotin, thiamine, MoCo, FeS clusters etc.), which is a more sensible solution given that there only one per certain type of enzyme. Some amino acids that are supplemented Nature cannot make with ease, in particular chlorination and fluorination reactions in Nature can be counted with one hand.
Homoserine, homocysteine, ornithine and aminoadipate.
E. coli already makes homoserine for methionine and threonine biosynthesis and homocysteine via homoserine for methionine. Ornithine is from arginine biosynthesis and was kicked out of the genetic code by it. Gram positive bacteria, which do not require diaminopimelate, make lysine via aminoadipate ("homoglutamate").Homoalanine.

The simplest novel amino acid is homoalanine (aminobutyrate). 2-ketobutyrate is produced during isoleucine biosynthesis, which if it were transaminated it would produce homoalanine. Therefore it is likely that the branched chain transaminase probably must go to some effort to not produce homoalanine (forbidden reaction). This amino acid is found in meteorites and is really simple, but its similarity to alanine, hence why it must have lost out.
Norvaline, norleucine and homonorleucine.
In the Weber and Miller paper the presence of branched chain amino acids was mentioned as potentially a result of the frozen accident. From a biochemical point of view, the synthesis of valine and isoleucine from pyruvate + pyruvate and ketobutyrate + pyruvate follows a simple pattern (decarboxylative aldol condensation, reduction, dehydration, transamination). The leucine branch is slightly different and is actually a duplication of some of the TCA cycle enzymes (Jensen 1976), specifcially it condenses ketoisovalerate (valine sans amine) and acetyl-CoA, dehydrates, rehydrates, reduces, decarboxylates and transaminates. If the latter route is used with ketobutyrate and acetyl-CoA one would get norvaline, with ketopentanoate (norvaline sans amine) and acetyl-CoA one would get norleucine. These biosynthetic pathway have actually been studied in the 80s. The interesting thing is that it can be used further making homonorleucine. Straight chain amino acids make sturdier protein, so there is a definite benefit there. The reason why norleucine is not in the genetic code is that it was evicted by methionine, which finds an additional use in SAM cofactor (Ferla and Patrick, 2014). Parenthetically, as a result the AUA isoleucine codon is unusal —it is also a rare codon (0.4%) in E. coli— as to avoid methionine has an unusual tRNA (ileX), which would make an easy target for recoding.Allonine.
Threonine synthase is the sole determinant of the chirality of threonine's second centre.Aminophenylanine.
Chorismate is rearranged to prephenate and then oxidatively decarboxylated and transaminated to make tyrosine, while the hydroxyl group of chorisate is swapped for amine for folate biosynthesis. If the product of the latter followed the tyrosine pathway one would aminophenylalanine.Rethinking phenylalanine.
 While on the topic, the whole route for phenylalanine biosynthesis is odd. It feels like an evolutionary remnant. If one were to draw up phenylalanine biosynthesis without knowing about the shikimate/chorismate pathway the solution would be different. If I were to design the phenylalanine pathway I would start with a tetraketide (terminal acetyl-CoA derived; 2,4,6,8-tetraoxononanoyl-CoA), cyclise (Aldol addition of C9 in enol form to C4 ketone), two rounds of reduction (6-oxo and 8-oxo) and three dehydrations (4,6,8-hydroxyl), followed by a transamination (2-oxo): phenylalanine by polyketide synthesis!
While on the topic, the whole route for phenylalanine biosynthesis is odd. It feels like an evolutionary remnant. If one were to draw up phenylalanine biosynthesis without knowing about the shikimate/chorismate pathway the solution would be different. If I were to design the phenylalanine pathway I would start with a tetraketide (terminal acetyl-CoA derived; 2,4,6,8-tetraoxononanoyl-CoA), cyclise (Aldol addition of C9 in enol form to C4 ketone), two rounds of reduction (6-oxo and 8-oxo) and three dehydrations (4,6,8-hydroxyl), followed by a transamination (2-oxo): phenylalanine by polyketide synthesis!Naphthylalanine.

Phenylalanine has a single aromatic ring, naphthylalanine has two. Using the logic for the rethought phenylalanine synthesis we get a synthesis by hexaketide (2,4,6,8,10,12-hexaoxotridecanoyl-CoA). Namely cyclise (Aldol addition of C11 in enol form to C6 ketone and C13 to C4), two rounds of reduction (8-oxo and 10 or 12-oxo) and five dehydrations (4,6,8,10,12-hydroxyl), followed by a transamination (2-oxo). Naphthylalanine has been added to the genetic code (Wang et al., 2002), but was added as a supplement. GFP doesn’t work with either form of naphthylalanine (Kajihara et al., 2005), therefore there isn’t a good selection marker where the amino acid itself is beneficial. So probably the worst sketched pathway to possibly make.
Other aromatic compounds.
Secondary metabolites are often made by polyketide or isoprenoid biosynthesis, which are rather flexible so some extra compounds could be drawn up.LEGO style.
All amino acid backbones are make in different ways and only cysteine, selenocysteine, homocysteine and tryptophan operate by a join-side-chain-on-with-backbone approach. Tryptophan synthase does this trick by aromatic electophilic substitution, where the electrophile is phosphopyridoxyl-dehydroalanine (serine on PLP after hydroxyl has left), while the sulfur/seleno amino acids are by the similar Micheal addition. So this trick could be extended to other aromatic, carbanions and enols, if one really wanted to, but that if far from a nice one size fits all approach.Wednesday, 6 May 2015
Speculations about methionine biosynthesis genes
Last year I wrote a review on the bacterial diversity methionine biosynthesis:
It was crammed with facts and a couple of deductions that in my opinion are correct. However, there were a lot of hypotheses and conjectures, from plausible to wild, that did not make it into paper. Here I thought I might mention a few.
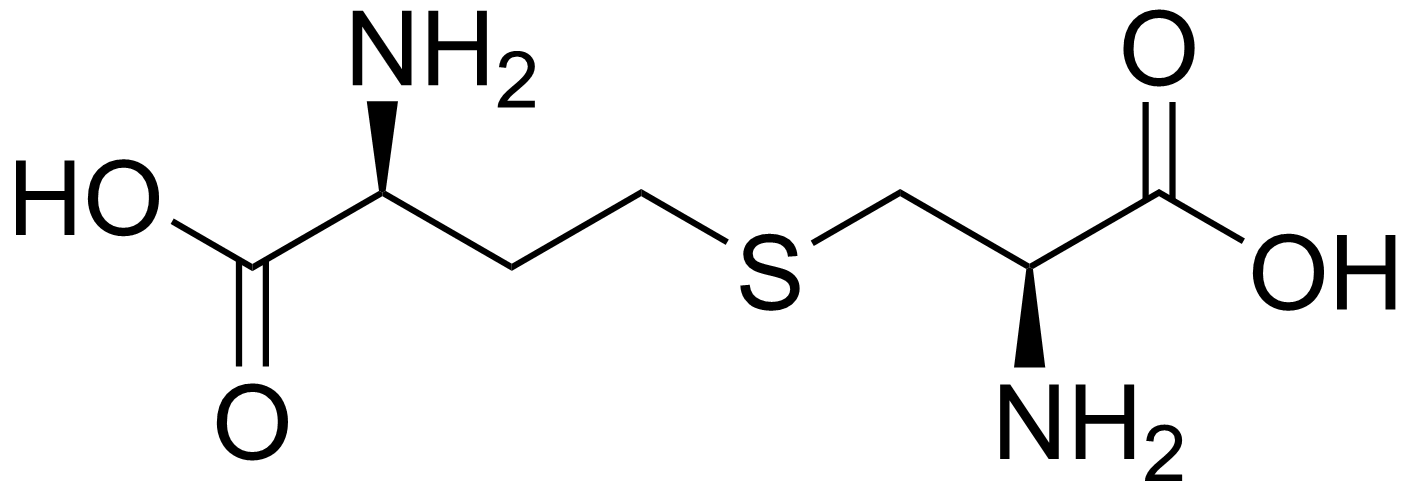 Cystathionine is a cysteine/alanine and a homocysteine/homoalanine joined together with a thioether. It has a short side (S is on the β) and a long side (S on the γ).
Cystathionine is a cysteine/alanine and a homocysteine/homoalanine joined together with a thioether. It has a short side (S is on the β) and a long side (S on the γ).
MetC is cysthationine β-lyase, it eliminates cystathionine at the thioether bond. On the shorter side (β).
MetB is cystathionine synthase it eliminates O-acetyl-homoserine at the ester bond and then attacks it with cysteine's thiol making cystathionine.
The two PLP enzymes hold cystathionine at some point but in radically different ways, one on the β side (MetC), the other on the γ (MetB).
Taking a step back, we have two types of cystathionine lyase and what controls the specificity between a β-lyase and a γ-lyase is not known —there have been a few papers looking into making MetC into a MetB and viceverse, but unfortunately nothing tackling this simpler issue. Cystathionine looks nearly identical from both sides: the sulfur bridge is hard to tell apart from a methyl group as there is only a slight size and charge difference. Methionine can be substituted with norleucine in protein with only minimal effect. Therefore it is intriguing how the enzymes bind it tightly in a specific way. My theory is that sulfur-π interactions may be involved as there are several tyrosines in the active site of MetC. Additionally, a β-elimination might be easier than a γ-elimination, therefore it is shame that there is a decent amount of data of the lack of γ-elimination activity in the β-lyase, but not viceversa. Therefore it would seem more likely to have a powerful bifunctional β- γ- cystathionine lyase than have retrained one that is strongly specific. However, this bifunctional enzyme is not an evolutionary a good idea due to the number of round trips it would do. Specifically, cysteine or homocysteine would go into making cystathionine, which the uncommitted lyase would either correctly transform or return a starting substrate —at the cost of ATP. The reason for this fascinating parenthesis is to conclude that cystathionine synthase/lyase combo that could do both, would be equally as bad of an idea —it would work due to flux from excess substrate to product in demand, but it is just extremely inefficient.
Ferla MP, Patrick WM. Bacterial methionine biosynthesis. Microbiology. 2014 Aug;160(Pt 8):1571-84.
PMID: 24939187, doi: 10.1099/mic.0.077826-0 and pdf.
PMID: 24939187, doi: 10.1099/mic.0.077826-0 and pdf.
The MetCombo
It is my opinion that a bifunctional enzyme that catalyses both the MetC and the MetB reaction is impossible. I have come to call this hypothetical enzyme, the MetCombo. So the data at hand are:- MetC and MetB are close homologues and it is really hard to tell them apart in a phylogram —with the bold assumption that the uncharacterised genes are what have been guess.
- Both KEGG and EcoCyc take the close homology to mean that bifunctional enzyme is present in several organisms —basically all those with MetB, which is a lot as you know from the met biosynthesis paper
- They are in the same pathway
- Nobody has ever seen a metCombo
- Papers that try to evolve MetC ↔ MetB are not realy successful
- Personal results: E. coli metC cannot rescue metB
- Personal results: Thermotoga maritima "metB" is actually a metC and it has no in vivo or in vivo MetB activity(check out my thesis)
- Catalytically a metC and metB in a single active site would be a disaster.
Catalytic profligacy
MetC is cysthationine β-lyase, it eliminates cystathionine at the thioether bond. On the shorter side (β).
MetB is cystathionine synthase it eliminates O-acetyl-homoserine at the ester bond and then attacks it with cysteine's thiol making cystathionine.
The two PLP enzymes hold cystathionine at some point but in radically different ways, one on the β side (MetC), the other on the γ (MetB).
Taking a step back, we have two types of cystathionine lyase and what controls the specificity between a β-lyase and a γ-lyase is not known —there have been a few papers looking into making MetC into a MetB and viceverse, but unfortunately nothing tackling this simpler issue. Cystathionine looks nearly identical from both sides: the sulfur bridge is hard to tell apart from a methyl group as there is only a slight size and charge difference. Methionine can be substituted with norleucine in protein with only minimal effect. Therefore it is intriguing how the enzymes bind it tightly in a specific way. My theory is that sulfur-π interactions may be involved as there are several tyrosines in the active site of MetC. Additionally, a β-elimination might be easier than a γ-elimination, therefore it is shame that there is a decent amount of data of the lack of γ-elimination activity in the β-lyase, but not viceversa. Therefore it would seem more likely to have a powerful bifunctional β- γ- cystathionine lyase than have retrained one that is strongly specific. However, this bifunctional enzyme is not an evolutionary a good idea due to the number of round trips it would do. Specifically, cysteine or homocysteine would go into making cystathionine, which the uncommitted lyase would either correctly transform or return a starting substrate —at the cost of ATP. The reason for this fascinating parenthesis is to conclude that cystathionine synthase/lyase combo that could do both, would be equally as bad of an idea —it would work due to flux from excess substrate to product in demand, but it is just extremely inefficient.
Conflicting results
Some methionine gene rescue experiments go in different ways that expected and there occasionally are concentration dependent oddities. My opinion is that this is due toone of the following:- It is dominating the threonine branch point and there is not enough threonine being produced.
- It is depleting all the cysteine or homocysteine
The ancestral PLP-dependent methionine gene
TBAMethionine and norleucine
TBAAlignment file
Here is the alignment file of manually aligned genes of various metB metC etc.Tuesday, 29 July 2014
Metabolic engineering Breaking Bad
Disclaimer: Obviously, this is a speculative what-if out of intellectual curiosity and by no means condones narcotics and their creation.
 |
| The logo of Breaking Bad had it been bio. |
Biocatalysis: greener and safer
Some time soon, biocatalysis and metabolic engineering will replace many heterocatalytic processes as it is more efficient, cheaper, safer and greener.Worldwide, there is a big problem of exploding methamphetamine labs. This problem could be fixed by switching from toxic and dangerous heterocatalysis processes to green and safe biocatalysis ones.
Requirements
So if Walt and Jessie wanted to win the green chemistry award, what would they need to do?Their major problem is the starting material and the production steps. So the whole lot. Therefore, they need to do metabolic engineering.
The major issue is that methamphetamine is not a natural compound, so extensive engineering would be needed to produce the final steps. However, once they laboriously made a production strain, they would need to set up a large-scale bioreaction, i.e. a brewing tank, extract the product by phase-separation, remove the solvent and purify by crystallisation as they normally do. Then the only worry then is that they may be as contaminant-prone as Hank is at brewing.
Starting point

The natural molecules most similar to methaphetamines are pseudoephedrine and ephedrine. These two diastereomers differ from the former in having a hydroxyl group (in different chiral orientations) on the carbon adjacent to the benzene ring. The biosynthetic pathway is known (PMID 22502775), but requires eleven steps, which have not been assembled exogenously in an orderly way.
Additionally, to convert ephedrine to methamphetamine new enzymes need to be engineered for the hydroxyl reduction, which is problematic. Consequently, ephedrine biosynthesis might not be the best route and instead something more radical may be in order.
Ephedrine biosynthesis
Ephedrine gets its name from the genus Ephedra, whose members produce it. Unfortunately the selective pressures for plant secondary metabolism are rather unusual (cool) and as a result the metabolic routes get a tad tortuous.If one were to forget how plants like to do things, one would guess a simple pathway with a similar logic to threonine biosynthesis be present. Namely, the carboxyl is twice reduced and the remaining hydroxyl is isomerised to the right place.

The final step (not pictured), the N-methylation, would be simply accomplished by a SAM-dependent methyltransferase and is the only part that is correct.
In the isomeration step, one might anticipate that an enamine-ketimine tautomerism followed by an attack by water might occur ruining the effort. However, the isomerisation of homserine to threonine is done via a PLP enzyme which holds the amine, so this isn't the problem.

The problem is plants like to make second metabolism in an OCD way, starting from specific compounds (e.g. geranyl-PP, farnesyl-PP, cinnamoyl-CoA, coumaroyl-CoA, malonyl-CoA and acetyl-CoA), preferably using decarboxylative condensation.
The ephedrine pathway is no exception and shares the beginning of the pathway with many other compounds Phenylanine > cinnamic acid > cinnaomyl-CoA>>benzoyl-CoA. Then the unique part is that the benzoyl-CoA is condensed with pyruvate, reduced and transaminated.
In reality, whereas the full pathway is known, the genes themselves are not, simply because nobody has sequenced E. sinica. Although a group in 2009 has gone to the effort of making yeast take up the DNA of E. glauca via ion implantation —no kidding around there!— and make ephedrine, but they did not sequence a few hybrids or similar, but instead did a lot of tedious work with primers (PMID: 19280123). Consequently, their experiment would need to be repeated, but with sequencing.
Once the various genes are cloned into E. coli, preferable into a strain that overproduces phenylalanine (eg. from PMID: 17880710), the pathway would be optimised, giving an ephedrine-producing strain.

The last step
The last step is the trickiest.After those few years of work are done, the hydroxyl needs to be removed. Biochemically, hydroxyl groups are normally removed in two steps, the hydroxyl group is removed without adding an electron pair to the molecule by a hydroxylase, therefore leaving a double-bonded carbon, which is then reduced by a reductase. In some rare cases, the hydroxyl is reduced away. The most famous example is ribonucleotide reductase. The mechanism is rather mental and ugly.
The dehydration route is a problematic option however. A modified 3-hydroxyacyl-ACP dehydrase and the enoyl-ACP reductase from the fatty acid biosynthetic pathway seem like good candidates. However, the dehydrated enolamine would spontaneously tautomerise and hydrolyse as mentioned above.
This might not be that catastrophic as the product would be phenylpropanone, which being similar to phenylpropane-dione, the product of the pyruvate-benzoyl-CoA condensation. It might be promiscuously transaminated again by the cathionine transaminase or by phenylamine transaminase.
Nevertheless it is an odd way of doing things.
The N-methylation must be done last as the product is slippery being so hydrophobic. Normally, biochemistry likes to put a handle to hold stuff like that, such as phosphates, CoA and glycosides. In this case, a N-glycosilation would be a good option. The best bet, however, would be to move the N-methylation step after the hydroxyl reduction, the methyltransferase cannot discern between the precursor for ephedrine or pseudoephedrine, so it is probably fairly accommodating towards amphetamine.
Crazy way
The carboxyl group needs to be replaced with a methyl group. This is not an option from a biochemists' perspective as C-C bonds cannot be made that easily, unless by condensation or transmethylation on aromatic structures. In a typical methyltransferase the methyl donor is SAM, while the acceptor is a nucleophile (Lewis base), such as an amine or a hydroxyl that has been deprotonated by a catalytic acid. From a technical point of view as far as I can tell there should not be anything forbidding a PLP and SAM dependent decarboxylative C-methylation. After a decarboxylation the negative charge is partially absorbed by the PLP (electron sink), leaving an nucleophilic secondary aldimine. The enzymatic reside that favoured the departure of the carboxyl group (say, catalytic lysine) might compete with the SAM though.
In the literature there is no sign of such a reaction: there is a decarboxylative O-methyltransferase (PMID: 22247507) and the various cases of SAM and PLP dependent enzymes, e.g. aminomutases, rely on SAM as a radical donor. There are some enzymes that point towards the possibility of such a C-methyltransferase, such as on an enol-ketone tautomerism (PMID: 5490210 and PMID: 17784761). Nevertheless, such an enzyme, if possible, would require a lot of work and luck to pull off. So the safer option might sound a lot longer, but has a higher chance of working...
Selection
A side question is how to select for better variants. To evolve a strain to make methamphetamine an way to select for one is obviously needed.The traditional way would be to assay for those compounds by HPLC, but this would mean that the variants would be screened laboriously (especially in light of the optimisation required).
An option for a high-throughput approach is to make a transcription factor that responds only to the substance needed, so that it activates a fluorescent reporter which can be selected by a FACS (eg. PMID: 22276138). Unfortunately, the targets of amphetamines and those of catecholamines are membrane receptors. So the phenylalanine binding TF, tyrR-econded, would be a good candidate for engineering.
Toxicity
The oral murine LD50 of methamphetamine is slightly higher than capsaicin (55 vs. 46 mg/kg), so it is probably non-toxic to bacteria and due to its hydrophobicity it can be phase-separated easily from the aqueous environment. So a least one bit would be straight-forward.Conclusion
Given the recombination and sequencing for gene identification, the many rounds of engineering and so forth, it would be five years if they are lucky. So metabolic engineering breaking bad would not start even at its fifth season...And it would be expensive to do and there is no guarantee that their strain would remain safe — copying Walter's formula was an issue, here it would require only a stolen tube.
In brief, it would actually be a real pain to do and take years to cobble together, so unfortunately, metabolic engineering cannot make Walter and Jessie more green and free of precursor woes...
Subscribe to:
Posts (Atom)