PyMOL will strip LINK entries from PDBs on saving while NGL obeys only CONECT entries in PBDs. An exception is PyRosetta: it behaves very nicely with disulfides, isopeptide bonds ( cf. repo of PyRosetta code from Keeble et al.) and other crosslinks —mostly. As a result I thought I'd add a note on how to add them in PyRosetta.
Covalents in PDB files
The easiest and most cheatsome way is adding a LINK (or SSBOND for disulfides) entry to the PDB file and opening it in PyRosetta, which will do its magic to add that crosslink:
LINK NZ LYS A 11 CD GLU A 34 1.33
SBOND CYS A 24 CYS A 52 2.06
Rosetta rightfully cares about official spacing so typing it will be trickly, but this is the major challenge. However, the question begs asking is what is this magic that happens behind the scenes?.
Basics
First, the basics. In computational biochemistry, the definition of residue includes nucleobases, ligands, ions and
water molecules. Each residue ought to abide by a preset topology. In Rosetta the topology is stored in a params file,
which is loaded as a residue type, the archetype of that residue. Each one with a unique 3-letter code —mostly three
letters. In Rosetta there's the global residue type set, which can be altered with the command line
argument -extra_fa_res, and mutable residues type sets that can be altered (vide intra). Additionally, a residue in
Rosetta can be "patched", for example the terminal amino acids in a peptide chain may have the NtermProteinFull
and CtermProteinFull patches applied: in the former there's an extra two protons on the backbone nitrogen and an
oxygen (named OXT) on the carboxyl carbon. In the case of lysine and glutamate, these are canonical amino acids and
accept the sidechain conjugation patch (`SidechainConjugation`). When a pdb file or pdb block is read termini are
added automatically by patching the terminal residues (unless disabled with `use_truncated_termini` command line
argument) and the covalent bond between residues added as directed in the LINK entry by patching them depending on the
type of interaction (such as SidechainConjugation).
pose.residue(1).annotated_name() # 'A[ALA:NtermProteinFull]'
There are many patches available. Some are alternate forms of residues, which are normally stored in PDB files as
alternate residues names, such as phosphoserine, which is SEP in PDB but S[SER:phosphorylated] in Rosetta —doing a
round trip will dump it as SEP.
Anatomy of a connection
In PyRosetta, every residues has none or more residue connections, which can be crosslinks, peptidic bonds etc. These
residue connections are numbered, but the polymer connections have an extra layer of syntax, wherein the N terminal
connection is called the "lower" and the C terminal connection is the "upper". There's probably a reason for this
nomenclature, but a way I remember it is that nitrogen has a lower atomic zahl than carbon (5 vs. 6) or that the lower
connects to a residue with a lower residue index.
A connection between two residues (either an amino acid or ligand) can be inspected as follows:
this_idx:int = 11 # the known residue
this_residue:pyrosetta.rosetta.core.conformation.Residue = pose.residue(this_idx)
number_connections:int = this_residue.n_current_residue_connections()
other_idx:int = this_residue.connected_residue_at_resconn(3) # residue connection no. 3 (see note below).
other_residue:pyrosetta.rosetta.core.conformation.Residue = pose.residue(other_idx)
other_atomno:int = this_residue.connect_atom(other_residue)
other_atomname:str = other_residue.atom_name(other_atomno)
this_atomno:int = other_residue.connect_atom(this_residue)
this_atomname:str = this_residue.atom_name(this_atomno)
distance:float = ( this_residue.xyz(this_atomname) - others_residue.xyz(other_atomname) ).norm()
print(this_idx, this_atomname, this_atomno, other_idx, other_atomname, this_atomno, distance)
An amino acid reside has lower and an upper connection (N-terminal and C-terminal connection) and if it's got a crosslink it will be the third connection (resconn). In the params file, this will be CONN3. CONN1 with a LOWER and UPPER will fail. Therefore, for an amino acid connection 3 will certainly be the non-polymer one.
As a test subject I will use segfaultin, a fictional protein that I never finished making with multiple troublesome parts, such an isopeptidic bond, cystine bond and a non-canonical amino acid, .
from rdkit_to_params import Params
import requests
p = Params.from_smiles('CCCCC(N*)C(*)=O', name='NLE')
p.PROPERTIES.append('ALIPHATIC')
p.PROPERTIES.append('HYDROPHOBIC')
rts = p.add_residuetype(pose)
pdbblock = requests.get('https://raw.githubusercontent.com/matteoferla/segfaultin/main/1ubq.iso.ss.sep.nle.pdb').text
assert 'LINK' in pdbblock
pyrosetta.rosetta.core.import_pose.pose_from_pdbstring(pose, pdbblock, rts, 'foo')
Parenthetically, RDKit calls a string with a PDB block in it a pdbblock, PyMOL a pdb_str and pyrosetta
a pdb_string, so the variable name choice tells something about one's loyalties!
Let's inspect what the residues call themselves:
pose.residue(1).annotated_name() # Z[NLE:NtermProteinFull]
pose.residue(2).annotated_name() # Q
pose.residue(24).annotated_name() # C[CYS:disulfide]
pose.residue(76).annotated_name() # G[GLY:CtermProteinFull]
pose.residue(100).annotated_name() # w[HOH]
This shows that a regular amino acid is just a letter, a non-canonical amino acid or ligand is Z/X/w and its three letter code, while the patched residues have colon and the patch name. This is important for mutations. So let's do a roundtrip:
First let's remove the bond by mutating to alanine.
I chose alanine because that is what is done in alanine scanning experiment in the lab,
but it could have been anything except the unpatched residues as that will have kept the coordinates.
There is a method in MutateResidue called .set_preserve_atom_coords which preserves the coordinates of atoms
with the same name, which can backfire and result in highly contorted residues.
for res in (11, 34):
pyrosetta.rosetta.protocols.simple_moves.MutateResidue(target=res, new_res='ALA').apply(pose)
assert 'LINK' not in ph.get_pdbstr(pose)
Now that we have a blank slate, let's add the bonds back, first by add the residues with the patch.
A SidechainConjugation patch with no connection will be just a regular residue
without that atom, ie. no protons or oxygens added.
pyrosetta.rosetta.protocols.simple_moves.MutateResidue(target=11, new_res='LYS:SidechainConjugation').apply(pose)
pyrosetta.rosetta.protocols.simple_moves.MutateResidue(target=34, new_res='GLU:SidechainConjugation').apply(pose)
These are not linked. To achieve this there are two possible commands:
pose.conformation().declare_chemical_bond(seqpos1=11, atom_name1=' NZ ', seqpos2=34, atom_name2=' CD ')
or
pyrosetta.rosetta.core.util.add_covalent_linkage(pose=pose, resA_pos=11, resB_pos=34, resA_At=7, resB_At=7, remove_hydrogens=True)
The latter will remove relevant hydrogens, create a new patch if not a normally linked residue and call the former function.
However, even though Rosetta thinks there's a covalent bond between these residues, the distance will be off.
assert 'LINK' in ph.get_pdbstr(pose)
print('distance: ', (pose.residue(11).xyz("CD") - pose.residue(34).xyz("CD")).norm())
To fix it, one can just do a FastRelax cycle, preferably in cartesian mode:
scorefxn=pyrosetta.create_score_function('ref2015_cart')
cycles=3
relax = pyrosetta.rosetta.protocols.relax.FastRelax(scorefxn, cycles)
movemap = pyrosetta.MoveMap()
idxs = pyrosetta.rosetta.utility.vector1_unsigned_long(2)
idxs[1] = 11
idxs[2] = 34
resi_sele = pyrosetta.rosetta.core.select.residue_selector.ResidueIndexSelector(idxs)
movemap.set_chi(allow_chi=resi_sele.apply(pose))
movemap.set_bb(False)
relax.set_movemap(movemap)
relax.minimize_bond_angles(True)
relax.minimize_bond_lengths(True)
relax.apply(pose)
Now we can have a gander, but with the caveat that NGL ignores LINK entries... as I said, crosslinks are tricky.
import nglview as nv
view = nv.show_rosetta(pose)
view.update_cartoon(smoothSheet=True)
view.add_hyperball('11 or 34')
view
Anatomy of a patch
Patches live in the database under database/chemical/residue_type_sets/fa_standard/patches/.
In PyRosetta there is a database folder, which can be accessed by navigating to its installation folder:
os.path.split(pyrosetta.__file__)[0]
Sidechain conjugation patches, for example, are added thanks to the file SidechainConjugation.txt.
A cool thing is that there can be two definitions loaded, so the original SidechainConjugation.txt
can stay. The easiest way to add a patch is from the command line.
Patches are controlled by the residuetypeset:
rts:pyrosetta.rosetta.core.chemical.PoseResidueTypeSet = pose.residue_type_set_for_pose()
or
params_paths = pyrosetta.rosetta.utility.vector1_string()
params_paths.extend([paramsfile])
resiset = pyrosetta.generate_nonstandard_residue_set(pose, params_paths)
or
FULL_ATOM_t = pyrosetta.rosetta.core.chemical.FULL_ATOM_t
rts:pyrosetta.rosetta.core.chemical.PoseResidueTypeSet = pose.conformation().modifiable_residue_type_set_for_conf(FULL_ATOM_t)
The first is the global one, the second allows the addition of params files, while the last is the modifiable version that allows shenanigans like adding a residuetype from a string with a params block:
rts = pose.conformation().modifiable_residue_type_set_for_conf(pyrosetta.rosetta.core.chemical.FULL_ATOM_t)
buffer = pyrosetta.rosetta.std.stringbuf(params_block)
stream = pyrosetta.rosetta.std.istream(buffer)
new = pyrosetta.rosetta.core.chemical.read_topology_file(stream,
name,
rts)
rts.add_base_residue_type(new)
pose.conformation().reset_residue_type_set_for_conf(rts)
Patches are stored as a vector given by the method .patches:
patches:pyrosetta.rosetta.utility.vector1_std_shared_ptr_const_core_chemical_Patch_t = rts.patches()
patch:pyrosetta.rosetta.core.chemical.Patch = patches[1]
vt:pyrosetta.rosetta.core.chemical.VariantType=patch.types()[1]
The variant type is an enum, whose members are listed here. We can check what patches apply to a give residue:
cys_rt:pyrosetta.rosetta.core.chemical.ResidueType = pose.residue_type_set_for_pose().get_base_types_name3('CYS')[1]
[(patch.name(), [vt.name for vt in patch.types()]) for patch in rts.patches() if patch.applies_to(cys_rt)]
which yields:
[('D', []),
('CtermProteinFull', ['FIRST_VARIANT']),
('NtermProteinFull', ['LOWER_TERMINUS_VARIANT']),
('N_Methylation', ['N_METHYLATION']),
('protein_cutpoint_upper', ['CUTPOINT_UPPER']),
('protein_cutpoint_lower', ['CUTPOINT_LOWER']),
('disulfide', ['DISULFIDE']),
('SidechainConjugation', ['SIDECHAIN_CONJUGATION']),
('VirtualMetalConjugation', ['VIRTUAL_METAL_CONJUGATION']),
('Virtual_Protein_SideChain', ['VIRTUAL_SIDE_CHAIN']),
('N_acetylated', ['N_ACETYLATION']),
('C_methylamidated', ['C_METHYLAMIDATION']),
('NtermProteinMethylated', ['METHYLATED_NTERM_VARIANT']),
('S-conjugated', ['SC_BRANCH_POINT']),
('Cterm_amidation', ['CTERM_AMIDATION']),
('Virtual_Residue', ['VIRTUAL_RESIDUE_VARIANT']),
('acetylated', ['ACETYLATION']),
('MethylatedCtermProteinFull', ['METHYLATED_CTERMINUS_VARIANT']),
('AcetylatedNtermProteinFull', ['ACETYLATED_NTERMINUS_VARIANT']),
('AcetylatedNtermConnectionProteinFull',
['ACETYLATED_NTERMINUS_CONNECTION_VARIANT']),
('DimethylatedCtermProteinFull', ['DIMETHYLATED_CTERMINUS_VARIANT']),
('hbs_pre', ['HBS_PRE']),
('hbs_post', ['HBS_POST']),
('a3b_hbs_pre', ['A3B_HBS_PRE']),
('a3b_hbs_post', ['A3B_HBS_POST']),
('oop_pre', ['OOP_PRE']),
('oop_post', ['OOP_POST']),
('triazolamerN', ['TRIAZOLAMERN']),
('triazolamerC', ['TRIAZOLAMERC']),
('ProteinReplsBB', ['REPLS_BB'])]
Note that pyrosetta.rosetta.core.chemical.VariantType.DEPROTONATED isn't there,
because that and pyrosetta.rosetta.core.chemical.VariantType.PROTONATED
and pyrosetta.rosetta.core.chemical.VariantType.ALTERNATIVE_PROTONATION
aren't for patches.
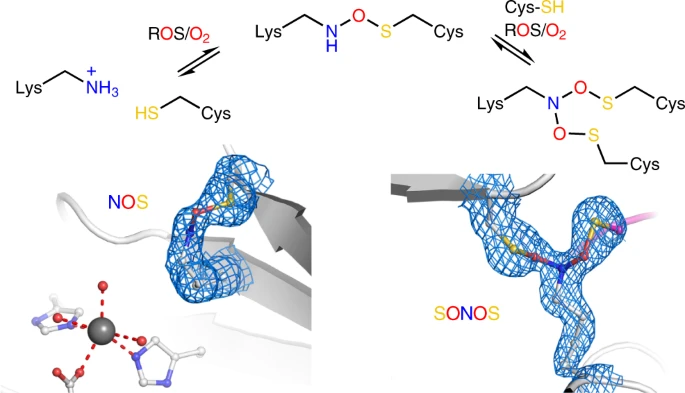
N-O-S bridge
In February this year (2022) a paper came out in Nature that showed that many protein had N-O-S bridges, a feature reported last year (2021, Nature). A N-O-S bridge is the sidechain conjugation of a lysine and hydroxycysteine, via the atoms nitrogen, oxygen and sulfur.
This would make for a nice example.
In the PDB file 6ZWJ from the latter paper is an example of this between lysine (LYS) 8 and hydroxycysteine (CSO) 38.
If we wanted to treat this properly,
we would need to add the patch SidechainConjugation for the residue CSO.
Making a residue CSO defined by a params file with CONN3 entry is also an option
but would be inelegant as it could not be used in isolation, even though CSO would not
be found in this context anyway.
Parenthetically, the oxidations of cysteine are S-oxycysteine (SCX),
S-nitrosocysteine (SNC), cysteine sulfenic acid (SDC), cysteinesulfonic acid (OCS)
So let's start by making a CSO residuetype and patch for it.
CSO.params:
NAME CSO
# *C(=O)[C@@]([H])(N(*)[H])C([H])([H])SO[H]
IO_STRING CSO Z
TYPE POLYMER
AA UNK
ATOM N Nbb X -0.2957777
ATOM CA CAbb X 0.0787753
ATOM C CObb X 0.1595662
ATOM O OCbb X -0.2971563
ATOM CB CH2 X 0.0420447
ATOM SG S X -0.0107892
ATOM OD OH X -0.3299774
ATOM H HNbb X 0.1237177
ATOM HA Hapo X 0.0557563
ATOM HB1 Hapo X 0.0425142
ATOM HB2 Hapo X 0.0425142
ATOM HD Hpol X 0.2284798
BOND CB CA
BOND CA C
BOND_TYPE C O 2
BOND CA N
BOND CB SG
BOND SG OD
BOND CB HB1
BOND CB HB2
BOND CA HA
BOND N H
BOND OD HD
PROPERTIES PROTEIN ALPHA_AA L_AA POLAR SC_ORBITALS
FIRST_SIDECHAIN_ATOM CB
BACKBONE_AA ALA
CHI 1 N CA CB SG
CHI 2 N CA C O
CHI 3 CA CB SG OD
CHI 4 CB SG OD HD
UPPER_CONNECT C
LOWER_CONNECT N
NBR_ATOM CA
NBR_RADIUS 4.556737904333845
ICOOR_INTERNAL N 0.000000 0.000000 0.000000 N CA C
ICOOR_INTERNAL CA 0.000000 180.000000 1.479082 N CA C
ICOOR_INTERNAL C 0.000000 69.099439 1.516005 CA N C
ICOOR_INTERNAL UPPER 55.959949 59.496115 1.552345 C CA N
ICOOR_INTERNAL O 173.362709 51.230560 1.218710 C CA UPPER
ICOOR_INTERNAL LOWER 66.923203 66.676171 1.468631 N CA C
ICOOR_INTERNAL CB 129.676916 70.236808 1.535445 CA C N
ICOOR_INTERNAL SG 169.561221 68.101623 1.828532 CB CA C
ICOOR_INTERNAL OD -177.727087 81.275795 1.662137 SG CB CA
ICOOR_INTERNAL H -123.392905 70.662367 1.024697 N CA LOWER
ICOOR_INTERNAL HA -113.868432 72.686858 1.099708 CA CB C
ICOOR_INTERNAL HB1 122.852027 67.860323 1.095516 CB CA SG
ICOOR_INTERNAL HB2 -118.751443 70.100055 1.096489 CB CA SG
ICOOR_INTERNAL HD -101.189326 68.731845 0.989399 OD SG CB
Patch:
NAME SidechainConjugation
TYPES SIDECHAIN_CONJUGATION
## general requirements for this patch
## require protein, ignore anything that's already nterm patched:
BEGIN_SELECTOR
NAME3 CSO # Add to this list as more sidechain-conjugable types are added.
NOT VARIANT_TYPE SC_BRANCH_POINT
NOT VARIANT_TYPE PROTONATED
NOT VARIANT_TYPE VIRTUAL_METAL_CONJUGATION
NOT VARIANT_TYPE SIDECHAIN_CONJUGATION
NOT VARIANT_TYPE TRIMETHYLATION
NOT VARIANT_TYPE DIMETHYLATION
NOT VARIANT_TYPE METHYLATION
NOT VARIANT_TYPE ACETYLATION
END_SELECTOR
BEGIN_CASE ## S-hydroxycysteine (CSO), L- or D-version #################################################
## These define which residues match this case:
BEGIN_SELECTOR
NAME3 CSO #L-version.
END_SELECTOR
# These are the operations involved:
# Delete a sidechain hydrogen:
DELETE_ATOM HD
# Add a sidechain connection and a virtual atom:
ADD_CONNECT OD ICOOR 180.0 75.00 1.793 OD SG CB
ADD_ATOM V1 VIRT VIRT 0.00
# Add new bonds:
ADD_BOND OD V1
# Set position of the new V1 atom:
SET_ICOOR V1 0.00 75.00 1.793 OD SG CONN%LASTCONN
REDEFINE_CHI 4 CB SG OD V1
END_CASE
Import them:
import pyrosetta
import pyrosetta_help as ph
pyrosetta.init('-in:file:extra_res_fa CSO.params -in:file:extra_patch_fa SidechainConjugation.txt')
Alternatively, if imported via options, do remember to register these with the relevant mover or residuetypeset.
patch_files = pyrosetta.rosetta.utility.vector1_std_string(1)
patch_files[1] = '...'
pyrosetta.rosetta.basic.options.set_file_vector_option('in:file:extra_patch_fa', patch_files)
pyrosetta.rosetta.protocols.simple_moves.MutateResidue(...).register_options()
Create a pose and link:
pose = pyrosetta.pose_from_sequence('AC[CSO:SidechainConjugation]AGGGK[LYS:SidechainConjugation]G')
print(pose.sequence())
CSO_idx = 2
CSO_atom_idx = pose.residue(CSO_idx).atom_index('OD')
LYS_idx = 7
LYS_atom_idx = pose.residue(LYS_idx).atom_index('NZ')
pyrosetta.rosetta.core.util.add_covalent_linkage(pose=pose,
resA_pos=CSO_idx,
resB_pos=LYS_idx,
resA_At=CSO_atom_idx,
resB_At=LYS_atom_idx,
remove_hydrogens=False)
assert 'LINK' in ph.get_pdbstr(pose)
minimise:
scorefxn=pyrosetta.create_score_function('ref2015_cart')
cycles=3
relax = pyrosetta.rosetta.protocols.relax.FastRelax(scorefxn, cycles)
movemap = pyrosetta.MoveMap()
movemap.set_chi(True)
movemap.set_bb(True)
movemap.set_jump(True)
relax.set_movemap(movemap)
relax.minimize_bond_angles(True)
relax.minimize_bond_lengths(True)
relax.apply(pose)
view:
import nglview as nv
view = nv.show_rosetta(pose)
view.add_representation('hyperball', f'{CSO_idx} or {LYS_idx}')
view.download_image('NOS.png')
view
No comments:
Post a Comment